
You are currently browsing the category archive for the ‘Pharmaceuticals’ category.
Prologue: What follows is a look at the use of 68Gallium as part of a positron emitting radioligand from an organometallic chemist’s point of view. I’m not from nuclear medicine nor am I a radiation oncologist.
It had to happen … the other shoe has dropped. My stage-4 prostate cancer has come charging back for round 2 after 9 years. Again, I’ve taken a personal interest in radiation oncology. Recently, my PSA shot up steeply through the 4.0 ng/dL threshold triggering an appointment with my radiation oncologist who has ordered a PET/CT scan. Back in 2015 I finished 18 months of hormone ablation (chemical castration) and got the PSA from 29 down to 0.01 with Lupron injections and earlier, a large cumulative dose of x-radiation in the lower abdomen. I have to say that while I experienced no discomfort at all in this round of treatment, I did lose body hair and muscle mass.
PET/CT scanning is an important tool in locating prostate cancer cells. Riding the platform in and out of the scanner is expensive but important. Unfortunately for me, the CT contrast agent is a potent emetic so the scanner becomes an expensive vomitorium ride.

The story of PET, Positron Emission Tomography, has evolved over decades of advancement. To begin, tomography, detectors and computers had to be invented. Separately, positron emission as a medically viable radiation source had to be identified and validated. A substrate for selective delivery of the isotope must be found. In the case of 18Fluorine, it is available as an organofluorine molecule like 18F-Glucose. It turns out that the 18F-Glucose concentrates in clinically useful places and K18F does not.
Positron Emitters
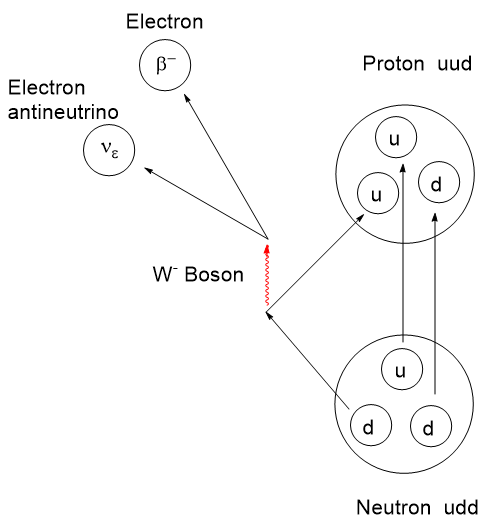
Atomic nuclei that are deficient in neutrons can have an instability leading to emission of a positron (anti-electron with a + charge), also called a β+ decay, which lessens the neutron deficiency by ejecting a positive charge from the nucleus. When a positron is ejected from the nucleus it finds itself immediately swarmed by the electron clouds of surrounding atoms and molecules and doesn’t travel very far. When a positron encounters a negatron (regular electron, β−), they annihilate one another and emit two gamma photons of 511 keV energy at 180 degrees apart. This is a mass to energy conversion. Loss of one positive charge from the nucleus gives rise to a transmutation of the atom causing a one-unit drop in atomic number, that is it goes from n+ to (n – 1)+, but retains most of its atomic weight. In this case, 6831Gallium undergoes positron decay to 6830Zinc.
Positron emitters include 11Carbon (T1⁄2 = 20.4 min), 13nitrogen (T1⁄2 = 10 min), 15oxygen (T1⁄2 = 2 min), 18fluorine (T1⁄2 = 110 min), 64copper, 68gallium, 78bromine, 82rubidium, 86yttrium, 89zirconium, 22sodium, 26aluminium, 40potassium, 83strontium, and 124iodine. This a list given by Wikipedia, but there are many more in more comprehensive tables.
The actual mechanism of β-type emission requires a venture into fundamental particles called quarks. Protons and neutrons are composite particles called hadrons, not fundamental particles. Protons and neutrons are each comprised of 3 quarks, but with a different combination of “up and down flavors” where flavor refers to the species of quark. There are 6 flavors of quarks: up, down, charm, strange, top, and bottom. Interconversion between protons and neutrons can occur if one of the 3 top or bottom quarks changes flavor. By all means, if this interests you, take a dive into it. I shall stop here.
Positron emitters tend to have a short radioactive half-life as well as a limited chemical half-life in the body before they are cleared out through the kidneys or other routes. Ideally, the goal is to deliver a high radiation dose selectively to a target tissue as fast as is safe then disappear. Prolonged irradiation to surrounding tissue is undesirable. The optimal radiopharmaceutical will be highly target selective and have a short half-life. A selective radiopharmaceutical is one that will accumulate in a desired cell type or organ. Accumulation can be aided through simple solubility, the ability to undergo transport through a cell wall, affinity to a specific receptor and the ability to function fast enough to resist the various clearance mechanisms.
A short half-life means that the radioactivity per gram of radioisotope, specific activity in Becquerels per gram, will be at its maximum after activation. Though the radioactivity may be intense, the radiation dose can be controlled by the amount of mass administered. With radioisotopes, there are two kinds of purity to consider: Chemical purity referring to the atoms and molecules present; Radiological purity referring to the presence or absence of other radioactive isotopes. To provide maximum safety and effectiveness, the specific radioisotope with the desired decay mode should be the only source present. If your desired source is an alpha emitter, you don’t need spurious quantities of a gamma emitter present because of inadequate purification.
Economical methods of preparing positron emitters had to be addressed. To fully exploit PET for any given situation, tissue selectivity of radioligands had to be determined and selective positron radiopharmaceuticals developed. Due to the short half-life of these radioisotopes, rapid and safe methodologies to produce them by efficient nuclear transformations, isotope isolation followed by chemical synthesis had to be developed. It is important that isotope generation, isolation and attachment to a ligand be done nearby the hospital for the proper activity to reach the patient.
Positron emitter production involves a nuclear reactor for neutron activation or a cyclotron accelerating protons or deuterons in the preparation. Because both of these sources are highly destructive to organic molecules, an inorganic radioisotope is produced separately and chemically modified to produce an inorganic species that can be chelated or otherwise attached to a radiopharmaceutical. This technique evolved from simple radiography in the 1930’s to a large array of techniques and applications today. The reader is invited to take a dive into this topic.
Since my cancer experience began, a few new radiotherapies and imaging agents have landed in oncology space for prostate cancer. Recently I posted on Pluvicto PSMA (Prostate Specific Membrane Antigen) which was before I knew about my current prostate situation. PSMA is a transmembrane protein present in prostatic cells. Pluvicto uses a chelated 177Lutetium beta emitter as the destructive warhead and a peptidomimetic fragment for binding to the PSMA receptor.
A Brief Interlude into Quality Factor
It should be noted that the various forms of particle (alpha, beta, or neutron) or electromagnetic radiation (x-ray or gamma) have differing abilities to penetrate and cause ionization of within matter. There is a factor for this which is used to refine dosage calculations. It is called the Quality factor, Q.
The destructive effects of radiation stem from its ability to ionize matter along its path. Ionization is a disruptive effect that may result in fragmentation of molecules or crystal lattices into reactive positive or negative ions. Single electron radical species may be formed as well. It is possible for some fraction of the disrupted molecules to recombine if the fragments haven’t already diffused away or gone on to further transformations.
The deleterious effects of radiation on living tissue stems from the amount of disruptive energy transferred to tissues along the path of each particle. Charged particles like electrons, protons and alpha particles tend to dump their energy into matter rapidly and along a short path making them less penetrating than neutrons or electromagnetic rays in general.
Quality factor, Q, is a dimensionless coefficient that is multiplied by an absorbed dose to give a more realistic estimation of radiation energy absorption. Interestingly, the Q for neutrons varies with energy and rises to a maximum around 0.5 to 1 MeV of energy and falls off at higher energies.
The larger the Q factor, the larger the corrected radiation effect. X-, gamma, and beta radiation have a Q factor lower than the others by a factor of 10 to 20. The x- and gamma rays will tend to pass through matter leaving a small amount of their energy to disruption. In radiation therapy this is compensated for by just increasing the fluence or the exposure time.

For clarity, x-rays are generated from the electron cloud around an atom via electron transitions. For instance, if an electron is dislodged from an inner, low energy orbital, another electron can occupy that vacancy by the emission of an x-ray. Gamma rays originate from nuclear energy transitions. Often a nuclear decay might result in a new nucleus that is not at its ground state and would be categorized as metastable. This metastable state, which has its own half-life, can collapse to its ground state by the emission of a gamma ray matching the loss of energy by the nucleus.
Neutrons
Free neutrons are special. They undergo beta decay with a short half-life outside the nucleus having t1/2 = ~ 10-15 minutes, depending on the information source. Not having a charge, they tend to be more penetrating than other particles. However, effective shielding can be had with a hydrocarbon like paraffin or water by virtue of the high concentration of hydrogen nuclei present in these substances. Neutrons are not affected by charge repulsion from an atomic nucleus and therefore can collide and interact with the hydrogen nucleus (a proton). They can scatter from hydrogen nuclei, leaving behind some of their kinetic energy with each collision (see “Neutron Lethargy“). This scattering is the basis for using water to moderate the neutrons in a nuclear reactor. Neutrons are cooled by repeated collisions with hydrogens in water to the point where their kinetic energy of 0.025 eV, which from the Maxwell-Boltzmann distribution corresponds to a temperature of 17 oC, thus the term “thermal neutrons“.
Many elements absorb neutrons, increasing the atomic weight and very often altering the stability of the nucleus leading to a radioactive decay cascade. This is what is happening in neutron activation. In the case of water, the ability of free neutrons to collide with hydrogen nuclei allows them to dislodge hydrogen ions or free radicals from organic and biomolecules resulting in ionization and makes them quite hazardous to living things.
Radioligands
Drugs like Pluvicto are referred to as a radioligand. There is a radioisotope connected to an organic “ligand” for selective binding to a specific protein receptor. A radioligand is injected and diffuses its way a particular receptor where it binds. As it turns out, due to the gamma radiation also emitted by 177Lu, Pluvicto is a radioligand that can also be located in the body by the gamma radiation it emits. In general, a radioligand can be used for two endpoints: To find and signal the location of a particular cell type; and to find and vigorously irradiate a particular cell type.
There are recent radioligand compounds that are used as PET (Positron Emission Tomography) diagnostic agents which selectively bind to the PSMA receptor where they can undergo positron emission revealing the site of prostate cancer cells by tomography. 18F-glucose was first synthesized in 1967 in Czechoslovakia at Charles University by Dr. Josef Pacák and was first tested as a radiotracer by Abass Alavi in 1976 at the University of Pennsylvania on volunteers. Positron tomography came along later. Cancer cells consume glucose faster than normal cells so the 18F will tend to accumulate to a slightly greater extent and reveal their position by positron annihilation. The two 511 keV x-rays simultaneously detected at 180o apart are identified by a ring coincidence detector. A single detection event is discarded.


A radioligand that received FDA approval the same day as Pluvicto was Locametz or Gallium (68Ga) gozetotide. This gallium radioligand targets PSMA as does Pluvicto but is only a PET diagnostic agent.
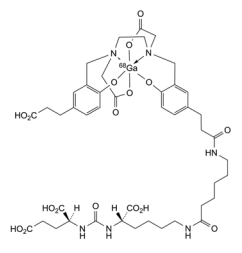
Locametz has 4 carboxylic acid groups, a urea group and two amide groups aiding water solubility and numerous sites for hydrogen bonding of this radioligand to the receptor. The organic portion of the Locametz is called gozetotide, named “acyclic radiometal chelator N,N’-bis [2-hydroxy-5-(carboxyethyl)-benzyl] ethylenediamine-N,N’-diacetic acid (HBED-CC).” The 68Ga (3+) cation is shown within an octahedral complex with a single hexadentate ligand wrapping around it. The short 68 minute half-life of 68Ga requires that a nuclear pharmacy be nearby to prepare it. The short half-life of 68Ga or other positron emitters as well as the possibility of destructive radiolysis to the ligand prevents preparing a large batch and stocking it. Locametz must be synthesized and transported prior to use. This rules out remote or rural hospitals.
Nuclear Chemistry
So, where does one obtain 68Gallium? Well, there are several methods out there. 68Ge/68Ga generators are produced commercially. One company is GeGantTM who offers 1-4 GBq of 68Ga. (Note: 1 GBq is 1,000,000,000 disintegrations per second).

From the scheme above we see the workings of a 68Ga generator. The ligand attachment is performed exterior to the generator. Atomic nuclei that are neutron deficient like 68Germanium can transform a proton to a neutron. There are two ways this can happen. In Electron Capture (EC) an inner “s” electron can be absorbed by a proton converting it to a neutron and emitting a neutrino by the weak nuclear force. This lowers the atomic number by 1, in this case 6832Germanium becomes 6831Gallium. The other mechanism is for the nucleus to emit a positron (anti-electron) and eject 1 positive charge as a positron (and an antineutrino) from the nucleus, resulting in a new neutron. The atomic weight remains constant, but the atomic number drops by one. If available energy in the nucleus is less than about 1 MeV, an electron capture is more favorable than positron emission.
Once you know about the 68Ge electron capture reaction leading to the 68Ga isotope you have to ask, where does the 68Germanium come from? There are a few different ways to make and concentrate 68Ge and the method you use depends on the equipment available to you. One way is to accelerate protons to a high energy in a cyclotron and slam them into atoms heavier than germanium, such as rubidium or molybdenum. The collision with break the target nuclei into pieces by a process called “spallation“.

Cyclotrons
The first cyclotron was independently invented by Ernest Lawrence 1929-1930 at UC Berkeley. It was the first cyclic particle accelerator built. The idea of the cyclic accelerator was first conceived by German physicist Max Steenbeck in 1927. In 1928-1929 Hungarian physicist Leo Szilard filed patent applications for a linear accelerator, cyclotron, and the betatron for accelerating electrons. Unfortunately for both Steenbeck and Szilard, their ideas were never published or patented so word of the ideas were never made public.
Where does one go to get a cyclotron? One company is Best Cyclotron Systems. If you are not sure of how a cyclotron works, check out the image below from Wikipedia. Note: A cyclotron can only accelerate charged particles like protons, electrons, deuterons and alpha particles which are introduced into the middle of the machine. A key component is the “D” or Dee, so-called because of their D-shape. The cyclotron has two hollow, coplanar Dees which are each connected to a high voltage radiofrequency generator. The Dees are open chamber-shaped electrodes that alternately cycle through positive and negative high voltage attracting and repelling charged particles under the influence of a powerful magnet. Because charged particles change their trajectory under the influence of a magnetic field, the particles follow a curved path of increasing diameter, accelerating until they exit the Dees and careen into the target.

The Drug Enforcement Agency (DEA) reports that it has seized xylazine and fentanyl mixtures in 48 of 50 states in the US. They mention that while fentanyl is responsive to naloxone, xylazine is not an opioid. Consequently, an overdose of a fentanyl/xylazine mixture may not respond to naloxone as expected.
DEA says that the great majority of fentanyl in the US comes from the Sinaloa and Jalisco cartels in Mexico, with the raw materials coming from China.
Xylazine is used in veterinary medicine as a sedative with analgesic and muscle relaxant properties. It would be interesting to see what the sulfur brings to the table as far as biological activity goes. You have to go to a bit of trouble to put the sulfur there so it must do something.


One reaction scheme for the preparation of xylazine is shown below. It is from expired US patent 4614798A (1985), Example 1, assigned to Vetamix. This prep uses acetic anhydride which is a List II chemical in the US. An acetamide intermediate is prepared from the aniline which is then reduced with sodium hydride and treated with carbon disulfide to give the reactive isothiocyanate intermediate. Finally, 3-aminopropanol adds to the isothiocyanate carbon to give the thiourea alcohol. Acid catalyzed cyclization by displacement of water by sulfur gives the xylazine product. This is some good meat-and-potatoes heterocyclic chemistry.

List II chemicals are subject to few serious reporting requirements in the US as seen in the table below. I suspect that the DEA benefits from good will disclosures. Following acetic anhydride shipment would be a good start in finding illicit labs in the US for several illegal drugs including xylazine, but it seems to be made in Mexico. Raw materials from China and drugs from Mexican labs add up to a very hard problem. Hard to tell how the cartel boys are making xylazine just from the internets.

First, a lot of chemists could say a lot of things on this topic. This is what I have to say. This essay is not written for medicinal chemists or molecular biologists. They already know this stuff. This is for everyone else. Learning usually requires an expansion of your vocabulary and this is no different.
When it comes to illicit drug synthesis I’ve always been a bit of a Puritan. As an organic chemist I’ve always felt that it is morally indefensible and a waste of talent for a chemist to make or help make dangerous and illegal drugs. Putting potent, illegal drugs on the market is like leaving a hand grenade in a playground.
For myself and for many others, what is fascinating about drug molecules is how structural features on the molecule confer pharmacological effects on biological systems. The molecular-level effects are referred to as a structure/activity relationship, or SAR. The chemical structure of the molecule makes all of the difference in how a drug works. What matters is the overall bulk, water solubility, acidity/basicity, hydrogen bonding and how charge is distributed on the molecule. As a reminder, in order for two molecules to react they must bump into one another in a particular way. And not just that, but bump into a particular spot oriented properly and with sufficient energy.
Drug molecules do not just swim directly to the site they are intended to go. They must take a random walk through flowing, jostling biofluid molecules and a minimum dose must survive the ordeal before they are metabolized, excreted or both. Some pharmaceuticals, called “pro-drugs“, are constructed in a way that relies on the action of metabolic processes to change them into the active drug. This is because they have some kind of chemical vulnerability and must be whisked into the body in disguise.
What the protein can do depends in large part on the sequence of amino acids that it is comprised of and how chain arranges itself. A protein polymer is made of a chain of amino acids that can interact with other molecules or with itself. Some lengths of a protein may lie flat and be somewhat rigid while other lengths may coil into a helical form. A protein molecule made of these features can then bunch up into a wad of protein that holds a particular shape. Along the surface of this shape are bumps, folds and crevices. In these places, there may be exposed amino acids that can attract acidic or positively charged parts of a molecule. Other spots may attract basic features like nitrogen with its lone pair of electrons. Still other places will attract molecular features with poor water solubility or just low polarity.
In order for a drug to act it must bump into the target biomolecule like a protein and at a particular site on the molecule. Some drugs may remain unchanged and just spend a lot of time stuck to a crevice (an active site) of an enzyme, preventing the intended biomolecule from doing so. Others may permanently bind to a protein or other molecule, thereby blocking it from doing its job. And others, like aspirin, may leave behind a fragment permanently blocking the active site of an enzyme. Some drugs prevent a protein or enzyme from working and are called antagonists. Others may activate it and are called agonists. What you aim for depends on the system you are trying to manipulate.
A molecule that binds with a protein is called a “ligand“. A ligand can connect with the protein through one or more attachment points. The greater the number and strength of the attachment point(s), the more time the ligand will stay attached. A ligand may even become permanently attached. Ligands purposely or externally provided for a desired outcome are considered as “drugs”. Ligands that cause an undesired outcome may be referred to as toxic. Not all ligands are aimed at human proteins, however, such as the beta-lactam antibiotics which bind with certain bacterial enzymes. This is a fascinating topic all by itself, but it is left as an exercise for the dear reader.
Ligand or drugs can have specific structural features that are associated with its activity or potency. This assembly of molecular features on the ligand is called a “pharmacophore“. An enzyme will have small region on its surface that can accommodate the “docking” of a ligand with the right shape and polarity. Different substances that share these features may comprise a family of substances having similar activity. In the case of opioids like fentanyl, this active site is referred to as an “opioid receptor“. There are a several variants of opioid receptors distributed throughout the human brain.
According to the DEA, fentanyl is the most serious drug threat the US has ever faced. In the 12 months ending January, 2022, there were 107,375 deaths from drug overdoses and poisonings. Of those, 67 % involved synthetic opioids like fentanyl.
Fentanyl is not found in nature. It is made in a reaction vessel or a bucket by a person. It is totally synthetic in origin and is prepared from other manufactured substances. The molecule is relatively simple and there are many places on it where new functional groups can be attached to produce designer analogs. Due to its startlingly high potency, a large number of doses can be made in fairly small batch equipment.
The explosion of fentanyl use is mind boggling. Drug cartels have taken to producing it themselves for greater profit and a more secure supply chain. The common syntheses are fairly simple, high yielding and, in the case of fentanyl, there are no stereochemical issues other than the atropisomerism of the amide bond. As far as purification goes, this isomerism is difficult to control and it is hard to believe that it is considered a problem by the “cooks” who make it.
A quick search of Google failed to bubble up information on what chemical form of illegal fentanyl commonly shows up on the street, whether as a free-base form or a salt. Like most amines, the free-base could be salted out of a reaction mixture by addition of an acid to a solution of free-base fentanyl in an organic solvent to produce the insoluble salt crystals. This solid material is then recovered by filtration. This is a common method of recovering amines from a reaction mixture.
It is worth looking at a synthesis of fentanyl to see what kind of chemistry is performed (see below). There is nothing remarkable about this synthesis- it’s just an example. A key raw material is the 4-piperidone hydrochloride on the upper left of the scheme. It is a piperidine derivative which is a feature of many drug substances. This one has 2 functionalities– the nitrogen and the C=O at the opposite end of the ring. Connections will be made at each end as the synthesis proceeds. The hydrochloride feature results from how the manufacturer chose to sell the product. Ammonium salts are frequently more shelf stable than the free amine.
The first step in the process below combines 4-piperidone hydrochloride with phenethyl bromide in the presence of cesium carbonate in solvent acetonitrile. In this transformation the nitrogen displaces the bromide to form a C-N bond connecting the fragments. Cesium carbonate is a base that scavenges acid protons. According to Wikipedia, cesium carbonate has a higher solubility in organic solvents than do the sodium or potassium analogs. Cesium carbonate is commonly used when a base stronger than sodium carbonate is needed. In order for the reaction to go forward, the HCl must be neutralized to liberate the free base. It is hard to imagine that the folks doing an illegal preparation are using a cesium base due to higher cost. The displacement of the bromide by nitrogen releases hydrobromic acid as well which must be removed from the mixture. Bromide is chosen because it is a good leaving-group. para-Toluenesulfonate, or tosylate, has been used as well.
Next, aniline must be added to the piperidone ring where the C=O is located. We have to end up with a single C-N bond connection from the aniline nitrogen to the C=O double bond then remove the oxygen and replace it with a hydrogen atom. Aniline is quite toxic and volatile with an LD50 of 195 mg/kg (dog, oral). It stinks too. This sequence is referred to as a “reductive amination“, meaning that the oxygen is replaced by single bonds to nitrogen and hydrogen. Adding hydrogen to a molecule is referred to as a reduction. The authors of the work commented that of three hydrogen donors tried, sodium triacetoxyborohydride gave the best yields. These borohydrides donate hydrogen as the negatively charged hydride, H:–.
Acetonitrile is a polar aprotic solvent that allows enough solubility to the reagents and intermediates so as to help the reaction along. Reductive amination classically proceeds through a C=N (imine) intermediate which then undergoes a hydrogen reduction of the bond to give the amine product.
The two-nitrogen intermediate is then fitted with a 3-carbon fragment bearing a C=O to the aniline nitrogen connected to the benzene ring. With this transformation, the amine nitrogen becomes an amide nitrogen. The fragment added is called propanoyl chloride (pro-PAN-oh-ill KLOR-ide) and involves the displacement of the chloride with the nitrogen producing hydrochloric acid. The purpose of the diisopropylethylamine base is to serve as an acid scavenger. The solvent was dichloromethane which is not uncommon for this kind of reaction. It has a low boiling point for easy removal by distillation and a slight polarity for dissolving substances that are somewhat polar. It is also inert to the reaction conditions.

It takes a high level of education, training and resources to design and perfect a process like the one above. However, it can be executed by most people after a bit of training. You don’t have to be a chemist to follow the procedure. The trick will be to avoid poisoning yourself from aniline or fentanyl exposure in the process.
However illegal fentanyl is made, the raw materials going into it must combine to give one unique final product. There are not an infinite number of pathways to fentanyl. However, structural variations of the raw materials could be chosen using the same basic reaction conditions to produce a spectrum of designer analogs. If specific molecules are outlawed, analogs can readily pop up to skirt regulations.
The people who make illicit fentanyl are sourcing the raw materials from somewhere. Unlike heroin, there are no natural substances in the manufacture of fentanyl. Heroin is just plant-based morphine that has been acetylated. Acetic anhydride is the choice commercial reagent for this. The acetic anhydride supply chain can be traced. Fentanyl, however, requires a supply chain for numerous fine chemicals. In the US, many substances are flagged by suppliers in a way that could cause the authorities to investigate the buyer. Furthermore, US commercial suppliers often could do a Dun & Bradstreet credit check on you to gauge your suitability as a customer. Commercial chemical suppliers will not ship to a residential address or PO box. So it takes a bit of business structure to get chemicals sent from established chemical suppliers to your address.
The way to avoid this hassle is to import from somewhere like Asia. Given the high potency of fentanyl, the mass of raw materials in a shipment could be very low. Most organic chemicals are whitish or colorless and can be mislabeled. The lower the molecular weight of a substance, the lower the mass that will be needed for the process. There are no high MW reagents in the scheme above.
Herein lies part of the problem with fentanyl. The raw materials have other uses in the chemical/pharmaceutical industry and these legitimate substances can received by unscrupulous operators who can repackage and divert shipments to the bad guys in countries along the Pacific coast of the Americas. It is just simple smuggling.
The estimated lethal dose of fentanyl for humans is 2 milligrams. According to one source, “The recommended serum concentration for analgesia is 1–2 ng/ml and for anesthesia it is 10–20 ng/ml. Blood concentrations of approximately 7 ng/ml or greater have been associated with fatalities where poly-substance use was involved.” Overdosing with fentanyl is reportedly treatable with naloxone. But this is only effective if your unconscious body is found by a sympathetic bystander and help is called in promptly. This is a very slender reed from which to hang your life.
It is left to the reader to look further into the pharmacology and therapeutic window details fentanyl. Suffice it to say that dosing yourself with illicit opioids is a stupidly risky business. The illegal opioid risk is multiplied by other additives or the possible presence of designer analogs which may be 10 to 100 times more potent. End-use safety is not a priority of those who make and distribute these opioids.
Given the estimated 2 mg lethal dosage, fentanyl should be regarded as a highly toxic substance. As long as there is demand for potent opioid substances, someone will provide it. When the oxycodone supply tightened recently, heroin demand rose. It’s a deadly whack-a-mole situation. The only answer is reduced demand.
I’m posting a link to the story of the astoundingly fast discovery and market entry of Pfizer’s Paxlovid, their small molecule contribution to the treatment of COVID-19. You have to wonder if the emergency use authorization came before the patents were allowed.

Recent Comments